
Trained with a joint understanding of protein and cell behavior, the model could help with diagnosing disease and developing new drugs.

Cambridge, MA – A protein located in the wrong part of a cell can contribute to several diseases, such as Alzheimer’s, cystic fibrosis, and cancer. But there are about 70,000 different proteins and protein variants in a single human cell, and since scientists can typically only test for a handful in one experiment, it is extremely costly and time-consuming to identify proteins’ locations manually.
A new generation of computational techniques seeks to streamline the process using machine-learning models that often leverage datasets containing thousands of proteins and their locations, measured across multiple cell lines. One of the largest such datasets is the Human Protein Atlas, which catalogs the subcellular behavior of over 13,000 proteins in more than 40 cell lines. But as enormous as it is, the Human Protein Atlas has only explored about 0.25 percent of all possible pairings of all proteins and cell lines within the database.
Now, researchers from MIT, Harvard University, and the Broad Institute of MIT and Harvard have developed a new computational approach that can efficiently explore the remaining uncharted space. Their method can predict the location of any protein in any human cell line, even when both protein and cell have never been tested before.
Their technique goes one step further than many AI-based methods by localizing a protein at the single-cell level, rather than as an averaged estimate across all the cells of a specific type. This single-cell localization could pinpoint a protein’s location in a specific cancer cell after treatment, for instance.
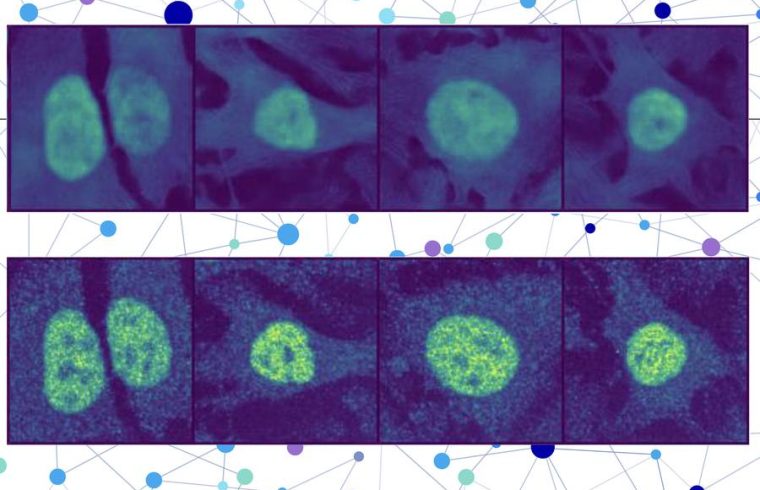
The researchers combined a protein language model with a special type of computer vision model to capture rich details about a protein and cell. In the end, the user receives an image of a cell with a highlighted portion indicating the model’s prediction of where the protein is located. Since a protein’s localization is indicative of its functional status, this technique could help researchers and clinicians more efficiently diagnose diseases or identify drug targets, while also enabling biologists to better understand how complex biological processes are related to protein localization.
“You could do these protein-localization experiments on a computer without having to touch any lab bench, hopefully saving yourself months of effort. While you would still need to verify the prediction, this technique could act like an initial screening of what to test for experimentally,” says Yitong Tseo, a graduate student in MIT’s Computational and Systems Biology program and co-lead author of a paper on this research.
Tseo is joined on the paper by co-lead author Xinyi Zhang, a graduate student in the Department of Electrical Engineering and Computer Science (EECS) and the Eric and Wendy Schmidt Center at the Broad Institute; Yunhao Bai of the Broad Institute; and senior authors Fei Chen, an assistant professor at Harvard and a member of the Broad Institute, and Caroline Uhler, the Andrew and Erna Viterbi Professor of Engineering in EECS and the MIT Institute for Data, Systems, and Society (IDSS), who is also director of the Eric and Wendy Schmidt Center and a researcher at MIT’s Laboratory for Information and Decision Systems (LIDS). The research appears today in Nature Methods.
Collaborating models
Many existing protein prediction models can only make predictions based on the protein and cell data on which they were trained or are unable to pinpoint a protein’s location within a single cell.
To overcome these limitations, the researchers created a two-part method for prediction of unseen proteins’ subcellular location, called PUPS.
The first part utilizes a protein sequence model to capture the localization-determining properties of a protein and its 3D structure based on the chain of amino acids that forms it.
The second part incorporates an image inpainting model, which is designed to fill in missing parts of an image. This computer vision model looks at three stained images of a cell to gather information about the state of that cell, such as its type, individual features, and whether it is under stress.
PUPS joins the representations created by each model to predict where the protein is located within a single cell, using an image decoder to output a highlighted image that shows the predicted location.
“Different cells within a cell line exhibit different characteristics, and our model is able to understand that nuance,” Tseo says.
A user inputs the sequence of amino acids that form the protein and three cell stain images — one for the nucleus, one for the microtubules, and one for the endoplasmic reticulum. Then PUPS does the rest.
A deeper understanding
The researchers employed a few tricks during the training process to teach PUPS how to combine information from each model in such a way that it can make an educated guess on the protein’s location, even if it hasn’t seen that protein before.
For instance, they assign the model a secondary task during training: to explicitly name the compartment of localization, like the cell nucleus. This is done alongside the primary inpainting task to help the model learn more effectively.
A good analogy might be a teacher who asks their students to draw all the parts of a flower in addition to writing their names. This extra step was found to help the model improve its general understanding of the possible cell compartments.
In addition, the fact that PUPS is trained on proteins and cell lines at the same time helps it develop a deeper understanding of where in a cell image proteins tend to localize.
PUPS can even understand, on its own, how different parts of a protein’s sequence contribute separately to its overall localization.
“Most other methods usually require you to have a stain of the protein first, so you’ve already seen it in your training data. Our approach is unique in that it can generalize across proteins and cell lines at the same time,” Zhang says.
Because PUPS can generalize to unseen proteins, it can capture changes in localization driven by unique protein mutations that aren’t included in the Human Protein Atlas.
The researchers verified that PUPS could predict the subcellular location of new proteins in unseen cell lines by conducting lab experiments and comparing the results. In addition, when compared to a baseline AI method, PUPS exhibited on average less prediction error across the proteins they tested.
In the future, the researchers want to enhance PUPS so the model can understand protein-protein interactions and make localization predictions for multiple proteins within a cell. In the longer term, they want to enable PUPS to make predictions in terms of living human tissue, rather than cultured cells.
This research is funded by the Eric and Wendy Schmidt Center at the Broad Institute, the National Institutes of Health, the National Science Foundation, the Burroughs Welcome Fund, the Searle Scholars Foundation, the Harvard Stem Cell Institute, the Merkin Institute, the Office of Naval Research, and the Department of Energy.





