
The researchers from Graduate School of Science, Osaka Metropolitan University, has investigated the specific computational conditions for Adiabatic State Preparation (ASP) executable on a quantum computer. ASP is considered one of the leading methods for efficiently taking into account electron correlation effects in the wave function of molecules.
This new research has clarified the computational conditions needed to use ASP, a major development for making quantum chemical calculations on a quantum computer practical for chemical or materials research.
ASP is a method to obtain the exact wave function by simulating the time evolution of the wave function under a time-dependent Hamiltonian. The Hamiltonian is an operator that defines all the relevant physical energies, such as the motion and interactions between electrons and nuclei that make up a molecule.
ASP gradually changes from a Hamiltonian whose eigenfunction is easy-to-prepare, to a true Hamiltonian during the quantum simulation of time evolution. However, the specific conditions for performing these calculations have not been investigated to a great extent, rendering their use impractical.
To make the method more versatile, the group of researchers including Dr. Kenji Sugisaki, Professor Kazunobu Sato, and Professor Emeritus Takeji Takui, have performed ASP numerical simulations under various conditions. As a result, the following four points have become clear:
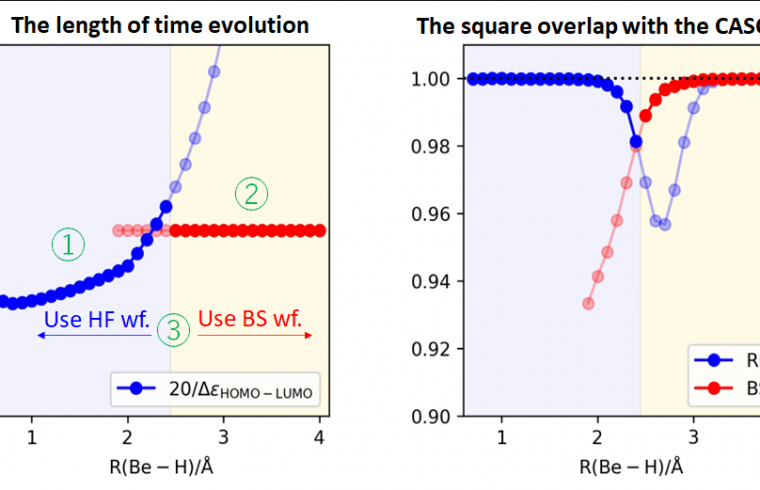
- When starting from the Hartree–Fock wave function, controlling the time variation of the Hamiltonian as sinusoidal functions gives the best performance.
- The amount of time to change the Hamiltonian should be inversely proportional to the HOMO–LUMO energy gap of a molecule under consideration.
- For electronic structures with open shell low-spin electronic configurations, a broken-symmetry wave function can be used to generate an accurate wave function with fewer steps.
- When using a broken-symmetry wave function, the length of time to change the Hamiltonian should be constant regardless of the molecular structure.

The research team has also proposed a simple method for determining whether the Hartree–Fock or broken-symmetry wave function should be used as an initial wave function.
The team has also demonstrated that these four points are relevant to the computational conditions for using ASP, making broader use practical for the first time.
“ASP is considered a powerful method for facilitating the preparation of correlated wave functions on a quantum computer, but the specific computational conditions that should be used have been unclear until now,” said Dr. Kenji Sugisaki.
“With this study, I believe ASP has eventually become practical. In the future, I expect this approach will be used to design new materials based on theoretical calculations using quantum computers”





