
Recent research reveals how one mutation causes fragile X, the most common inherited intellectual disability.

“Fragile X syndrome has been studied as a model of intellectual disability because in theory it’s comparatively simple,” says senior author Xinyu Zhao, a professor of neuroscience in the Waisman Center at the University of Wisconsin-Madison.
Fragile X patients have difficulty in learning and language, as well as temper tantrums, hyperactivity and extreme sensitivity to light and sound. The fragile X gene is located on the X chromosome and its mutation affects about 1 in 4,000 boys and 1 in 7,000 girls.
Nearly half of fragile X patients are also diagnosed with autism.
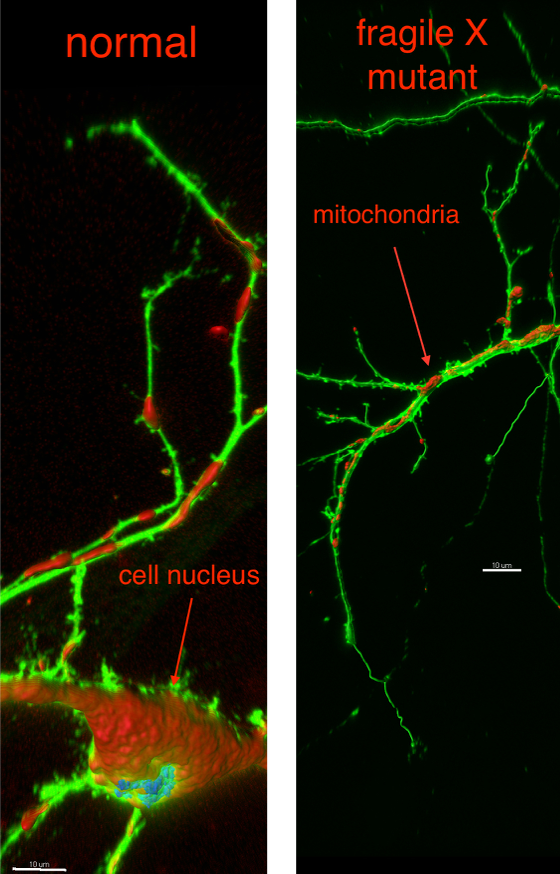
Zhao’s new study, in the journal Nature Neuroscience, shows that when mutated, the fragile X gene fails to produce its unique protein. As a result, subcellular units called mitochondria inside developing neurons are malformed, so the neuron is unable to create the necessary network of branches and contacts it needs to communicate.
These mechanisms link the fragile X mutation to the profound intellectual deficits in fragile X syndrome, Zhao says.
By blocking the proper formation and function of mitochondria, the fragile X mutation may also play a role in several other conditions. About 2 to 3 percent of people with autism have fragile X syndrome. “Autism is linked to more than 1,000 genes,” she says. “Because the fragile X gene is linked to more cases of autism than any other gene, what we have learned from fragile X helps us to understand autism.”
Despite the strong overlap, “until now, we did not know why this mutated gene causes either condition,” Zhao says.
Mitochondria have long been known as a cell’s powerhouse, but their other roles in brain development are only gradually becoming clear. Healthy mitochondria allow neurons to create more energy and have greater electrical activity. Mitochondria also support the establishment of dendrites, axons and synapses, parts of the elaborate linkages that allow brain cells to communicate with each other.
“Although mitochondria play a role in many diseases,” Zhao says, “this is the first time mitochondria dysfunction has been directly implicated in fragile X syndrome.”
Minjie Shen, lead author and a postdoctoral fellow in Zhao’s group, also transplanted immature, human neurons into mouse brains. Because these cells were grown from cells donated by fragile X patients, they carried the fragile X mutation. In these mice, low levels of the fragile X protein, FMRP, were associated with high levels of damaging oxidative stress caused by a variety of oxygen-bearing molecules.
“This is the first time that human fragile X neurons have been studied in any living brain,” Zhao says. “And so this information is more relevant to human neural development than what we can see in lab dishes.”
In a compelling demonstration of mitochondria’s role in fragile X symptoms, Zhao’s research group used a chemical that promotes mitochondria formation to reverse behaviors like hyperactivity and impaired social interaction in these mice.
The fragile X gene gains its power because it is a “regulator gene” that controls the action of dozens, even hundreds, of “downstream genes.” Indeed, the fragile X protein affects about 4 percent of messenger RNAs – compounds that “read” the genetic template of DNA to pattern new proteins.
The central finding of the new study, Zhao says, “is that we have discovered the first convincing mechanism that could explain the neurological impairment in fragile X, and that mechanism is defective mitochondria.”
Other developmental disorders, including Huntington’s disease, Rett syndrome and Down syndrome, seem to feature mitochondria dysfunction as well, she notes.
Mitochondrial mating: Function follows form
After a neuron distinguishes itself from its parent cells, the hundreds or thousands of mitochondria within engage in an elaborate dance, joining and separating in a dynamic balance that is vital to many biological functions. But with the fragile X mutation, “we see that the mitochondria are more fragmented, shorter and round rather than long and tubular, due either to decreased fusion or increased fission,” Zhao says.
The result is a neuron with impaired connectivity and less resistance to destructive oxidant chemicals.
“When we restored mitochondria fusion with gene editing or a chemical compound, we partly restored neuronal development,” Zhao says. “In mice lacking FMRP, we also rescued some behavioral deficits using the chemical treatment.
“This is the first direct evidence that mitochondrial dysfunction contributes to pathogenesis of fragile X,” she adds, “and I hope it will open new investigations and new therapeutic developments.”
Pinpointing a mechanism for fragile X is a first step to finding chemicals that might block or reverse that mechanism, Zhao says. Any treatment that is developed for these defects could be used after birth, during the period when neurons start to mature, Zhao says.
Although such a potential treatment is years away, the current study is a major advance for conditions that today lack treatments, Zhao says. “Human neuroscience seems to get more complex all the time, but I feel we’ve established a foothold that allows us to see the true source of difficulty in several serious neurological disorders. And that’s exactly our role as basic neuroscientists.”





